

 脸书专页
脸书专页 粉丝交流群
粉丝交流群MIT发布迄今最完整的新冠病毒基因注释图谱…
你听说过不需要科学实验,而是基于现有数据或者图谱,仅仅只是进行分析比对,就能得出相当精准结果的研究方法吗?
日前,麻省理工学院(MIT)研究团队宣布绘制出了迄今为止最完整的新冠病毒基因注释图谱。他们在通过比较大量相似病毒基因组后,确认了新冠病毒(SARS-CoV-2)基因组中未被发现的六个蛋白质编码基因。
相关论文于5月11日发表在《自然・通讯》(Nature Communications)期刊上,通讯作者是麻省理工学院计算机科学马诺利斯·凯利斯(Manolis Kellis)教授。
凯利斯在接受媒体采访时表示:“我们能够使用这种功能强大的比较基因组学方法进行进化签名,以发现这一极其重要的基因组的真正功能性蛋白质编码内容。”
西湖大学助理研究员鄢仁鸿对 DeepTech表示:“该研究主要采用大数据类比分析的研究手段,同时,他们自己有一套完善的验证程序,来对新冠病毒的突变做了系统性的归类研究,这对后续摸清病毒的进化规律具有重要的参考意义。”
 图丨相关论文(来源:Nature Communications)
图丨相关论文(来源:Nature Communications)
新的功能性蛋白编码基因被发现
新冠病毒属于严重急性呼吸综合征相关的冠状病毒,同时也是沙贝病毒(Sarbecovirus)的亚属病毒,这类病毒其实与人类早有接触,相似病毒也比较多。比如,导致2003年非典爆发的 SARS-CoV,双方便属于同一物种。
 图|新型冠状病毒(来源:Pixabay)
图|新型冠状病毒(来源:Pixabay)
在这种情况下,凯利斯团队选择通过比较基因组学来对这些相似病毒进行研究,比较基因组学是基于基因组图谱和测序基础上,对已知的基因和基因组结构进行比较的学科。
凯利斯此前也曾通过此类分析的计算技术来判断物种之间某些 DNA或 RNA碱基是否保守,并比较它们随时间的进化模式。另外,该团队还使用该技术将人类基因组与其他哺乳动物的基因组进行了比较,分析模式较为成熟。
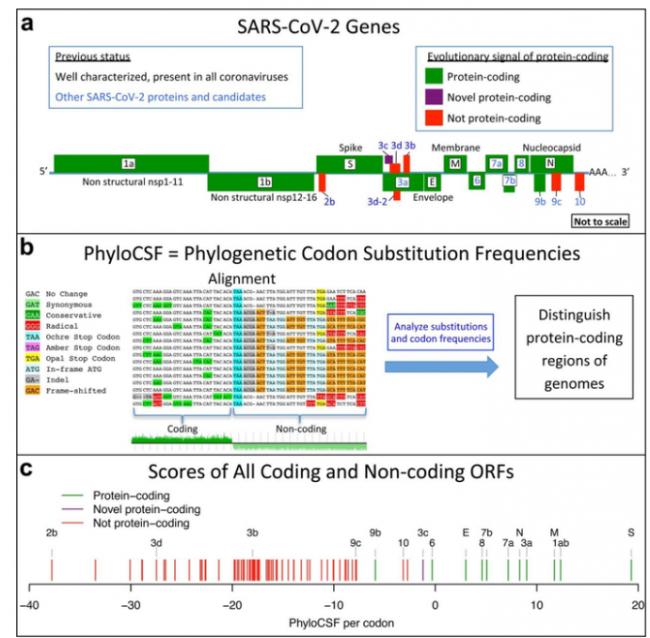 图|研究团队使用44株 Sarbecovirus株的比较基因组学来解析新冠病毒蛋白编码基因集(来源:Nature Communications)
图|研究团队使用44株 Sarbecovirus株的比较基因组学来解析新冠病毒蛋白编码基因集(来源:Nature Communications)
在比较基因组学的理论支撑下,MIT研究团队选择了44种 Sarbecovirus基因组,对蛋白质编码的进化特征和重迭约束进行了量化分析,以区分那些具有重要功能性的突变。
研究员对这44个紧密相关且完整的冠状病毒基因组和生成全基因组做比对,以评估蛋白质编码和核苷酸水平的限制,并注释同义受约束的密码子。最终确认了新冠病毒基因组中的六个蛋白质编码基因,并认为其他五个已经提出的不保守的基因区域不太可能编码功能蛋白。
 图|重迭3a的新基因3c编码蛋白质(来源:Nature Communications)
图|重迭3a的新基因3c编码蛋白质(来源:Nature Communications)
另外,研究团队还有一个新发现,即编码为 ORF3a的基因所在区域内还有另一个基因,科研人员将其命名为 ORF3c,该基因的 RNA碱基与 ORF3a重迭,但却有着不同的阅读框。这种基因有一个奇特的属性,它很少出现在大型基因组中,却又是病毒里的常客,不过目前这个新冠病毒新基因的作用尚不清楚。
鄢仁鸿认为,这项研究比较突出的亮点在于,它发现了一些之前被认为是新冠病毒编码基因的区域,并没有编码基因,而有一些被认为不会编码基因的序列,其实很可能是蛋白质的编码基因,从而帮助我们了解一些之前被忽略的蛋白在新冠疾病方面的具体功能。
一位来自复旦大学生命科学学院遗传学研究所的教授对 DeepTech表示,MIT研究团队通过比较基因组学取得了新的研究成果,在生物信息的分析比对上有自己的独到之处,即便这并不是一个新的研究方法。
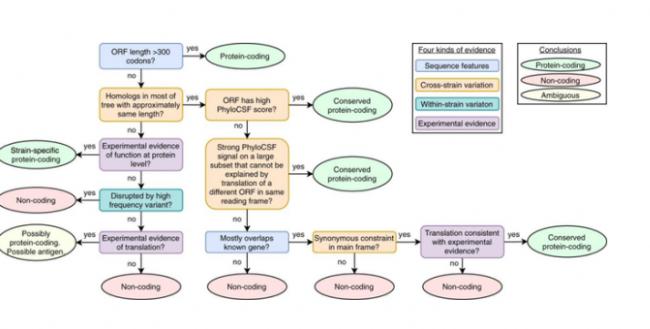 图|使用多步骤决策过程来区分功能性蛋白质编码基因(来源:Nature Communications)
图|使用多步骤决策过程来区分功能性蛋白质编码基因(来源:Nature Communications)
研究人员还发现,由于基因名称的不正确和相互矛盾,研究结果的准确性会出现许多波折,因此研究人员提出了命名新冠病毒基因的建议,并将这个建议发表在《病毒学》的另一篇论文中。
前文的复旦大学教授表示:“这个建议其实很有现实意义,不过在学术界,研究者往往有自己的个性及意愿,建议很好,但想要实现基因统一命名难度并不小。”
新研究大大推动探索病毒突变进程
在这项新研究中,研究人员还分析了自首次发现新冠病毒以来出现的1800多个突变。事实上,病毒异变往往会使病毒更具攻击性,从而导致人类的免疫系统很难发挥原有作用,感染性也会因此大幅提升,而此项研究恰好能在病毒突变方面给到精准评估。
复旦大学教授认为,这项研究分析了近2000个突变,而且也发现了一些帮助基因逃避免疫系统的关键蛋白,因此得出了突变可以产生免疫耐受作用的分析。“也就是说,这些在人群中传播的突变,之所以能够存活下来,说明是有免疫耐受功能的。这种基于生物信息的研究,就启发性来说作用是相当大的。”
不过不同于前面进行的相似病毒间的比较,这次比较的是突变基因在大流行开始前后的进化速度。
一般情况下,在当前的疫情大流行之前,长期快速进化的基因会继续进化,而缓慢进化的基因则会保持原有趋势。但现在,研究人员发现了一种新模式,这说明它已经适应了新的人类宿主。凯利斯认为,这种发现有助于探索病毒的进化原理。
同样有变化的是围绕病毒遗传物质的核衣壳蛋白区域,研究人员发现该区域的突变远超预期,这和原有的历史进化模式大相径庭。
 图|突变病毒逃避免疫系统(来源:Pixabay)
图|突变病毒逃避免疫系统(来源:Pixabay)
凯利斯说:“新冠病毒整个基因组中变化最大的区域就是这种核衣壳蛋白的中间位置。我们推测那些在该区域不会突变的变异体会被人类免疫系统识别并消除,而那些在该区域随机积累突变的变异体,实际上能够更好地逃避人类免疫系统并保持循环。”
变体中出现的突变同样引起了研究人员的关注,例如来自英格兰的 B.1.1.7菌株、巴西的 P.1菌株和来自南非的 B.1.351菌株,这些变体中出现了许多更多危险的突变,正是在这些突变的帮助下,病毒才能从容避开免疫系统。更严重的是,这些变体中的每一个一般还会携带其他突变。
“这些变体中的每一个都有20多个其他突变,因此,重要的是要知道其中哪些可能在做某事,哪些却没有。”该研究的主要作者、MIT计算机科学与人工智能实验室(CSAIL)科学家欧文・荣格里斯(Irwin Jungreis)说道。这时,比较基因组学中的数据就派上了用场。
研究人员介绍,这些数据可以帮助其他科学家将注意力集中,全力对付那些看起来最有可能对病毒的感染力产生重大影响的突变上。当然,如果你有其他方面的研究需求,同样可以使用这些带注释的基因图集,他们目前在加利福尼亚大学圣克鲁斯分校的基因浏览器上提供了相关服务。
谈及该研究可提升的空间,鄢仁鸿表示:“研究的局限在于,这类分析缺少功能实验数据的支持,得出的结果并不一定准确,还需要反复的验证,只能说是给出参考意见。”
研究结果尚缺乏实验数据支持
迄今为止,人类与新冠病毒的对抗已经持续了很久,药物和疫苗【小编推荐:显微镜学家发表对四家疫苗公司的成分分析】的研发仍然是解决疫情的重中之重。而且由于新冠病毒的突变性,科学家对新冠病毒基因组的填补过程一直存在巨大阻碍,在这种情况下,新冠病毒蛋白质编码图集的完善无异于一场及时雨,对破解病毒密码起着至关重要的作用。
不过我们也要注意到,在该研究中,沙贝病毒属下的所有已知病毒均属于与严重急性呼吸系统综合征相关的冠状病毒。因此,这项在目前已知的在沙贝病毒中可行的分析结果,可能并不适用于将来发现的其他沙贝病毒物种。
“有一点我们需要注意,这项研究主要是依据现有数据比对,但生物信息和实际情况一般差距很大,基因组学比对未必能够保证其准确度,归根结底还是需要大量实验数据的支撑,不过它由此带来的启发性完全值得肯定。”复旦大学教授谈道。
而且,对于新冠病毒的研究,我们的目光不能仅仅局限于新冠病毒基因组蛋白质编码身上,它的 RNA结构同样重要。但是这一点,很多人都会忽视,科学家们也不能例外,即使是 RNA在研究感染和疾病方面有着突出的表现。
 图| RNA如同大脑指挥而不负责具体执行(来源:Pixabay)
图| RNA如同大脑指挥而不负责具体执行(来源:Pixabay)
通常我们认为,病毒的遗传信息会先编码一些蛋白质,然后通过这些蛋白质来进行具体执行,而遗传信息就是 RNA,它像一个大脑,并不具备执行功能,这些功能一般都是依靠它编码的蛋白质来实现的。
对此,鄢仁鸿向 DeepTech解释,“病毒的遗传物质是 RNA,相比于 DNA来说,并没有那么稳定,因此在侵染过程中,可能会产生一些突变。比如,在复制自己自己遗传信息的时候就容易有一些错配,或者说点突变,这种点突变的概率是很低的,但是由于病毒复制基数比较大,因此可能会筛选出一些更具侵染性的病毒突变体。”
就像病毒和蛋白质的关系一样,一方发布指令,一方严格执行。科学家们在进行科研工作时同样需要做到这一点,既要有基于数据的理论分析,开创新思路,同时也要有大量的后期实验做佐证。
尤其对于新冠病毒(中共病毒)这样的病毒来说,相关的基因组图集总是处在突变与补充的变化过程中。这也就要求科学家们需要从更加全面的角度考虑问题,通过不断更新实验数据,最终一步步走近科学真相。
- 🔥免费PC翻墙、安卓VPN翻墙APP
- 🔥灵魂之谜|中华文化|治国大道
来源:深科技
